6Calling CNVs from Low-pass Genome Sequencing Data
Low-pass genome sequencing or low-resolution genome sequencing has been proposed as a cost-effective alternative to detect clinically significant copy number variations.
Compared to traditional genome sequencing at above 30x coverage, low-pass genome sequencing only needs to achieve 0.1-10x coverage depending on its requirement for the resolution of CNV detection.
Low-pass genome sequencing enables labs to have different tiers of assays to achieve various clinical targets. For example, if you wanted to totally cover deletion and duplication events as well as loss of heterozygosity, a minimum of 5x coverage is needed and this would only allow detection of very large (>20MB) LOH events. If you’re only looking for larger CNVs, such as aneuploidy, you can get by with even 0.1x coverage.
In addition, Low-pass CNV detection can be used to identify CNVs that are difficult to detect using other methods—for example, in identifying rare deletions in the genome.
Dr. Guo sees low-pass sequencing continuing to emerge as a primary alternative to microarrays given the often equal or better performance and non-biased analysis.
“I do see low-pass sequencing becoming a more popular alternative to microarrays. For labs looking to call CNVs that don’t have a microarray platform, but do have a sequencing platform, I would strongly suggest they consider low-pass sequencing. In our experience, the performance and resolution are equal to arrays—and sometimes even better. With arrays, your sensitivity is limited to where your probes are located. Low-pass sequencing is basically the same platform as genome sequencing. Rather than 40x coverage, you’re running 5x or 8x coverage—trying to catch the larger CNVs. It’s also important to note that shallow sequencing is PCR-free which means it is non-biased sequencing. It’s uniform data laid out across the entire genome, which isn’t limited to certain regions like with microarray. You may be able to detect events you otherwise would have missed because of probe limitation.”
— Dr. Fen Guo, Ph.D., FACMG, FCCMG, Clinical Laboratory Director at PerkinElmer Genomics
Watch our free webinar—
Genome sequencing reveals cause of multi-generational split hand/split foot with long bone deficiency—to see how Dr. Raymond C. Caylor, Assistant Director, Molecular Diagnostic Laboratory at Greenwood Genetic Center, utilized genome sequencing and Bionano’s N
xClinical software, to provide a diagnosis for a multi-generational family with split hand/split foot with long bone deficiency.
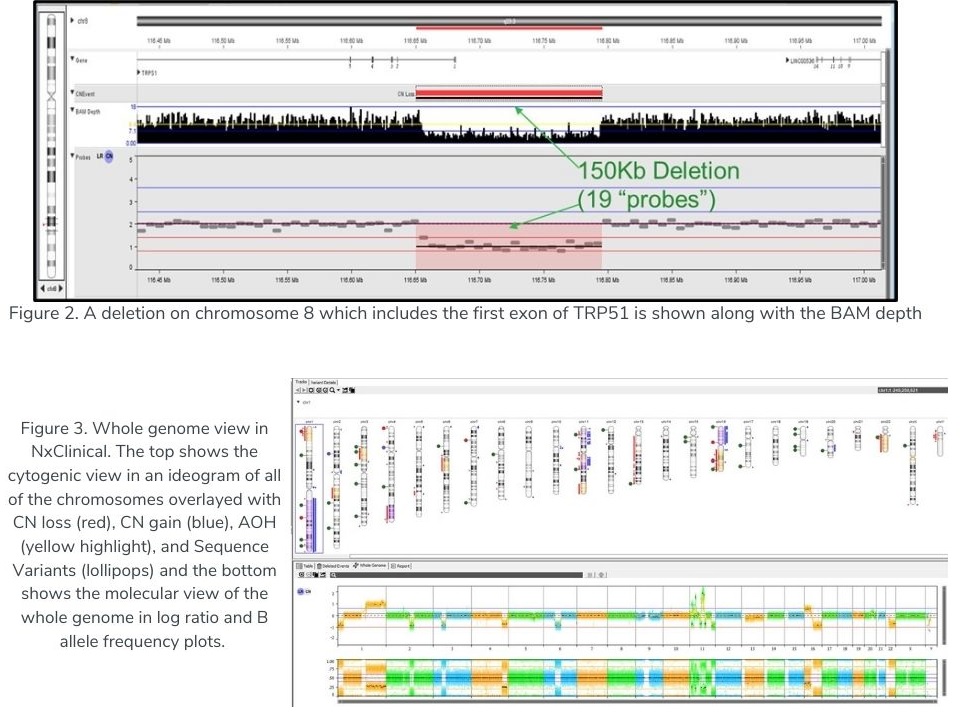 As described in
As described in 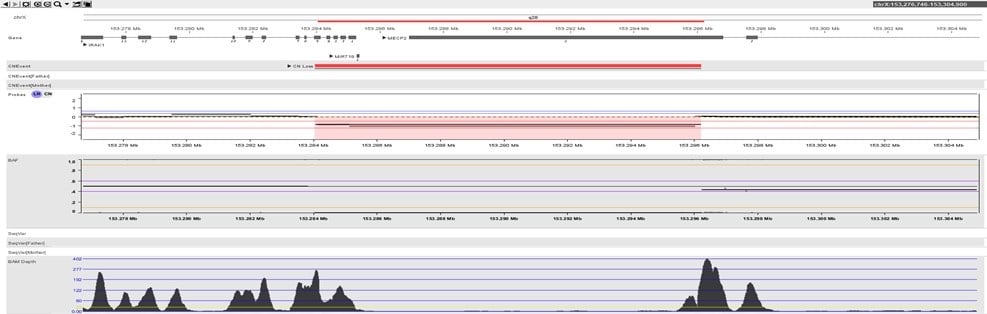
.jpg?width=996&height=204&name=Untitled%20(1).jpg)
.jpg?width=1430&height=356&name=Untitled%20(2).jpg)