Image Source: Copy number variation: Characteristics, evolutionary and pathological aspects (Biomedical Journal, 2021)
Open image in new tab
While anomalous structural changes have been observed in genes stretching back to the early 20th century, the modern concept of a copy number variant originated in the early 2000s.
As we mentioned before, most researchers until this time had seen genomic variation in humans through the lens of SNPs. But researchers like Charles Lee (currently with the Jackson Laboratory for Genomic Medicine), Michael Wigler (Cold Spring Harbor), and Stephen Scherer (Hospital for Sick Children, Toronto) were among the first to notice and document more genomic variation in the form of copy number variants.
As exhaustively laid out by Ingrid Lobo in a 2008 article published in Nature Education, it was Charles Lee who, in 2002, following a series of unsuccessful attempts to genotype patients, appears to have been the first to document observations “that healthy control patients showed major variations in their genetic sequences, with some having more copies of specific genes than others.” Lee then collaborated with Steven Scherer, “who had made similar observations, and together their labs used array-based comparative genomic hybridization approaches to measure the occurrence of these copy variants across the genome.”2
“Meanwhile,” Lobo explains, “Michael Wigler was also observing differences in copy numbers in healthy individuals using a complementary microarray technique involving representational oligonucleotide probes to detect amplifications and deletions in the genome.”
In 2004, these researchers published findings that “indicated large-scale variations in copy number were common and occurred in hundreds of places in the human genome, including areas coding for disease-related genes.”
BioDiscovery’s own VP of Sales, Dan Clutter, Ph.D., was fortunate enough to be on the frontline of CNV discovery at this time—helping equip researchers with microarray technologies that facilitated many of the initial studies into these variations.
Back then, when you looked at the genome at a molecular level, you expected to see two copies of every gene. But we started seeing variations in genes. At the time, most people talked about SNPs and changes in individual nucleotides.
When we saw changes in copy number, we were really surprised. And when we looked at cancer samples, we saw that variations were everywhere. Where you’d expect to see two copies of every gene, you were actually seeing giant pieces of DNA moving, disappearing, and amplifying. Then we came across labs doing treatments of various cancers with chemicals, compounds, anti-cancer compounds, and we would see the genome start to become normal again. It was astonishing.
In the early days of CV research, this was all done in cell cultures. It was a long time before we started sequencing human tumors to understand what was going on at a genomic level. Now, we’re doing single-cell sequencing, looking at a whole tumor and sequencing thousands of cells from that tumor. Some cells aren’t that disturbed. Others are. The evolution of CNV research has been truly fascinating.”
— Dan Clutter, VP of Sales, BioDiscovery
It would be roughly a decade from its discovery before CNV detection and analysis would be meaningfully applied in a clinical setting. During this time and since, thousands of publications marked a slow-moving explosion in our understanding of CNVs as researchers reveal more about their functions in human biology and usefulness in the clinic.
Below, we’ve excerpted a portion of the historical timeline presented in a recent review published in Biomedical Journal outlining the major milestones in CNV discovery and research. We’ve also compiled a few of the publications that mark these milestones through time.
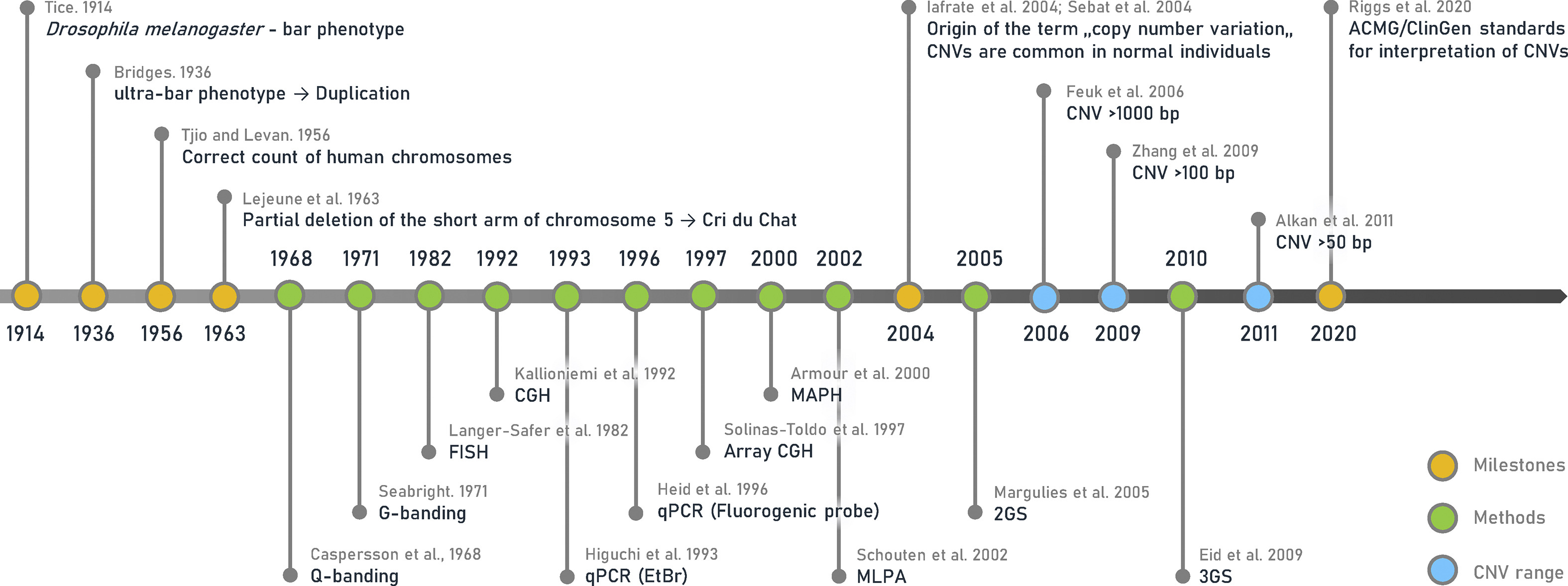
Image Source: Copy number variation: Characteristics, evolutionary and pathological aspects (Biomedical Journal, 2021)
Open image in new tab
Research into CNVs has produced many lines of evidence showing CNVs of certain genes are involved in how many types of cancer originate, develop, and progress—specifically through the alterations of their gene expression levels on individual or several cancer types.3 Clinically significant alterations can range from nucleotide-level insertions to deletions to entire chromosomes.
Shao et al. summarize some of the most notable research suggesting associations between CNVs and gene expression in cancers:
When I describe what cancer is, I try to get the point across that it’s really not one disease—it’s thousands of diseases. And in fact, it may be different for every single person. Our biology’s background mutational differences dictate what happens to new mutations—whether they’re effective or not and so on. It’s a complex and fascinating field, and it’s getting to a point now where people really understand the value at the clinical level, and they’re doing tests to look at diagnosis, prognosis, and so on.”
— Dan Clutter, VP of Sales, BioDiscovery
Other studies explored CNV and differential gene expression of several classical oncogenes or tumor suppressor genes in neuroblastoma,8 colorectal,9 and many other cancers.10 Detecting and analyzing CNVs has become central to diagnosing cancer at the molecular level. Research has shown that recurring deletions are typically overrepresented in tumor suppressor genes and underrepresented in oncogenes,11 and gene copy number aberrations (CNAs) may reveal therapeutic targets or markers of drug resistance in several types of cancer.12
Today, many clinical labs at the forefront of genomic capabilities sequence patient samples based on CNVs rather than SNPs to determine clonal evolutions—particularly the set of mutations that dominate a cell, which can often then be used to treat a disease. When looking at the copy number changes in a healthy person, one might see less than a handful of them. But the millions of SNPs present within every individual make it comparatively difficult to determine which are important for a particular disease.
Since the discovery of CNVs, researchers have revealed and documented a vast amount of CNA data generated by molecular-cytogenetic and genome sequencing-based methods.
This data has proved critical for identifying cancer-related genes and promoting further research into the relation between CNAs and various types of cancer. Most of this research has been focused on the association of CNA to common driver genes.13
A recent article published in Frontiers in Genetics offers a comprehensive background of CNV profiling for cancer and presents CNA signatures for 31 cancer subtypes. A complete list is included in the article’s Supplementary Signatures.
Here’s a sample of three common cancer subtype CNV signatures presented in this research:
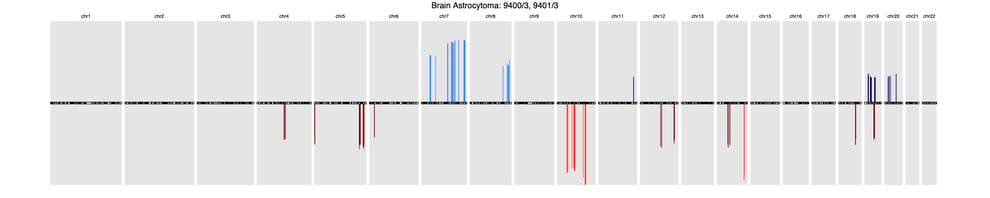
BrainBrain astrocytoma (9400/3, 9401/3)
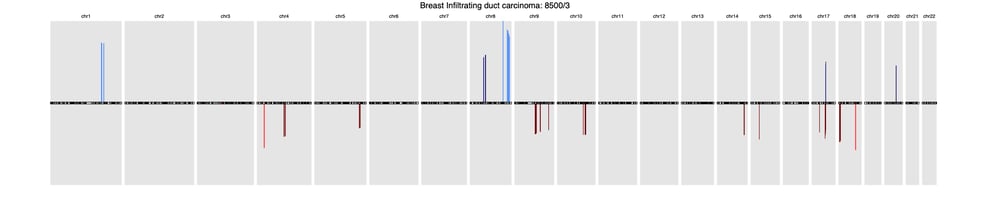
BreastBreast Infiltrating duct carcinoma (8500/3)
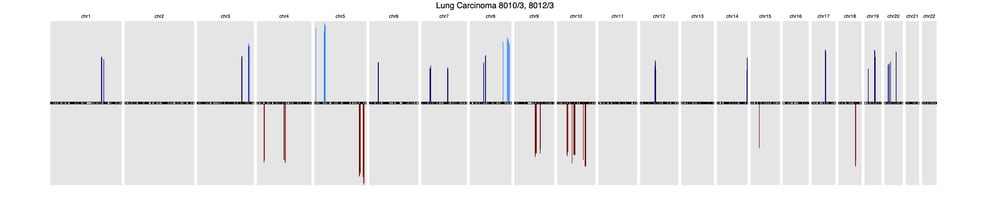
LungLung carcinoma (8013/3, 8012/3)
Accurately detecting, analyzing, and interpreting CNVs has become a routine and process in investigating tumor cells and diagnosing tumor patients.
Here at BioDiscovery, for example, we’ve built software that compares patient samples against the global CNV signature databases as well as a proprietary library of hundreds of different cancers to return the closest matches.
Our platform integrates many external databases to aid with interpretation, databases include OMIM*, DECIPHER, ClinGen (Prenatal, Postnatal, Dosage Sensitive), CIViC, Segmental Duplications, and more.
Learn more about our platform and its capabilities here.
Absence of heterozygosity (AOH) is another genetic characteristic known to cause genetic disorders through autosomal recessive or imprinting mechanisms—making it another hallmark of diseases including cancer.
“When looking at B-Allele Frequency (BAF) in the allele profile of the normal genome, you’ll find three bands: AA, AB, and BB. If an AOH region is present, you won’t see a copy number loss or change. You won’t know it was there, except that you lose the AB, and it becomes homozygous. So the BAF plot becomes really clear that there are only two alleles when there should be three. It turns out that this is also one of the signals of certain diseases and cancer is one of them.”
— Dan Clutter, Ph.D., VP of Sales, BioDiscovery
Here at BioDiscovery, we often work with labs focusing on AOH for cancer detection. The signatures link to certain cancers can also be found in clinical databases, which our software can conveniently and efficiently query.
Moscasism is another type of aberration that, when analyzed and interpreted accurately, can provide clinical insights into cancer.
“In cancer, it’s very difficult to get a pure tumor sample. Just about every time you excise a tumor, the sample will contain some normal tissue. So, a lot of the samples we look at have mosaicism; there are multiple cell types present rather than a single cell type. That’s another area of focus for many of the cancer labs we work with. They know certain things are mosaic, and certain things shouldn’t be. It becomes another hallmark of cancer and our platform can quickly unlock those insights through a convenient interface.”
— Dan Clutter, Ph.D., VP of Sales, BioDiscovery
Our platform has helped many researchers to detect and interpret the profiles from cancer samples where contamination by normal cells is quite common.
It’s important to note, however, that mosaic events can also appear as the clonal expansion of acquired post-zygotic mutations. Compared to constitutional defects in the same regions, mosaic abnormalities can result in milder phenotypes but they may also appear in otherwise seemingly healthy individuals.
Homologous recombination is one of the major mechanisms of defective DNA repair and frequently occurs in cancer. It’s emerging as both a promising biomarker and treatment vector for some types of disease. The genomic scarring often left by homologous recombination—commonly referred to as homologous recombination deficiency (HRD)—is now the subject of intense research and discussion to understand and reach a consensus on what information HRD contains as it pertains to a current or potential disease state.
While multiple consortiums attempt to harmonize and develop a standard for quantifying or scoring HRD, it appears that many in the field have accepted an HRD scoring model put forward in a 2020 article published in the Journal of Clinical Oncology.
This model establishes, for example, how many CNVs, breakpoints, and areas of AOH there may be in a patient sample with HRD and combine this data to arrive at an HRD score—much like in quantifying minimal residual disease (MRD) or tumor mutational burden (TMB).
NxClinical currently calculates HRD scores using a rules-based decision tree model to classify and quantify Genomic Scarring in the form of Loss of Heterozygosity (HRD-LOH), Telomeric Allelic Imbalance (TAI), and Large-Scale State Transition (LST). NxClinical's HRDScore is the sum of all of these Genomic Scars.
Learn more on HRD scoring in our 3-minute explainer video below here.
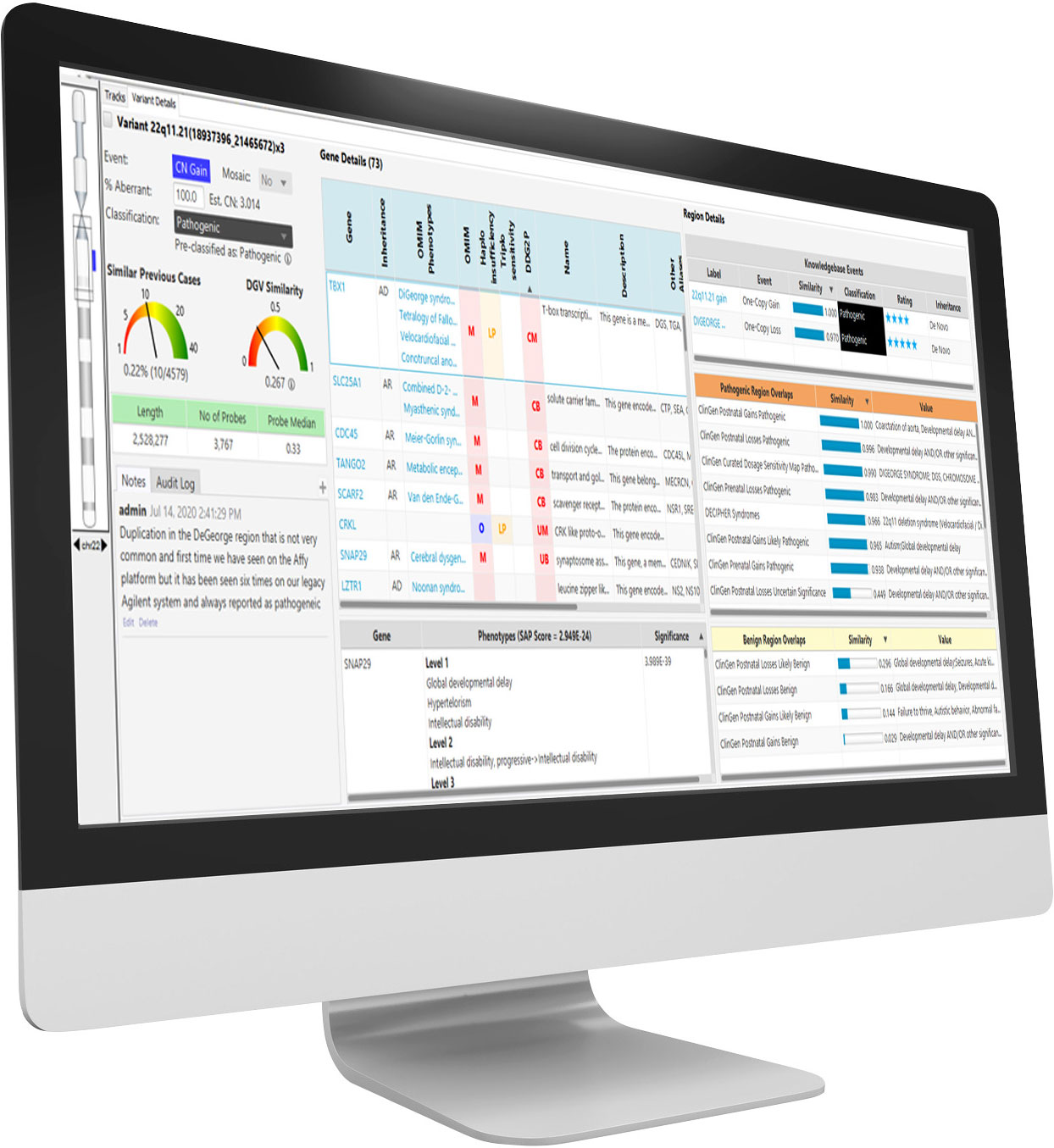
NxClinical is the most comprehensive single-software cytogenetics and molecular genetics solution for analyzing and interpreting CNV, SNV, and AOH data across all platforms for patient samples.
BioDiscovery’s CNV calling algorithms are the gold standard in the field for deriving CNVs from microarrays and NGS. BioDiscovery’s MSR algorithm makes it possible to obtain copy number from a variety of NGS data (WES, WGS, targeted panels, shallow sequencing). This allows clinical labs to get the most out of a single NGS assay—copy number, AOH, and sequence variants from the same assay streamlines the process and saves time and money.
Here are just a few of the reasons renowned clinical labs put NxClinical at the center of their cancer workflows:
Your lab—and the patients who rely on it—shouldn’t be stymied by constant software switching and unassisted sample interpretation. NxClinical detects, analyses, and visualizes genomic variants in one place. Simplify your clinical toolset while strengthening it with automation, AI, and variant prioritization tools that trim the list of potentially causative variants from hundreds or thousands to a handful. Make more informed decisions faster with fewer tools.
Clinical labs have long-deserved a genomic variant analysis tool that lets them fully utilize the data they already have. NxClinical enables you to finally extract the valuable CNV data from all your previous cases (yes, all of them) and compare them to your new ones. Automatically build a case history of analyzed samples and put it to work to continually improve your classifications.
Freeware isn’t free. Labs pay for it through a lack of capabilities, support, and updates—not to mention the inefficiencies they impose day after day. NxClinical delivers a level of value that far exceeds its cost. Unlike freeware’s lackluster (and often absent) visualization capabilities, NxClinical presents all the important information your lab needs to evaluate an event together on a single screen—unlocking new opportunities for quick and confident variant interpretation. Close-knit support and regular updates offer far more than any freeware can provide.